When you do a Bayesian inference, Markov chain Monte Carlo (MCMC) sampling is a common method to obtain a posterior probability of your model or parameter. So far, I have avoided using MCMC in my programs because I like simple and rapid algorithms. But, MCMC now looks unavoidable when I do more sophisticated Bayesian modeling.
There are many software platforms to do MCMC with reasonable learning curves, like Stan & Rstan or BUGS. These are definitely worth studying when you want to be a serious Bayesian.
But, for now, I choose a Python package called “PyMC“. This is because it is well integrated with Python language, and there is a nice introductory textbook to learn this package. (It is freely available online, and they sell hard copies too.)
After reading some chapters of the book, I tried to solve a simple problem with PyMC, which is related to phylogenetic inference and species delimitation.
The problem is estimation of λ in the model below.
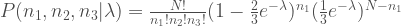
Why is this related to delimitation? Because this is a model of distribution of tree topology when you sample 3 individuals from 3 species.
If you sample 3 individuals from 3 different species and reconstruct gene trees, you are more likely to see one particular topology, the same one as species tree, than others. On the other hand, if you sample 3 individuals from 1 species, 3 types of topology are evenly observed. Finding this skew / evenness of topology distribution is a basic idea of the tr2-delimitation.
N in the model above is the number of sampled loci, which is a sum of n1, n2 and n3, counts of three possible tree topology of 3 samples. As λ (a branch length of species tree) increases, you more frequently observe topology 1 (species tree topology). The distribution becomes more even when λ is close to zero.
With this model, a posterior distribution of λ when you observe topology counts [n1,n2,n3] is,

I tried to estimate this distribution by MCMC. Luckily, there is an analytical solution to posterior distribution of λ at least with a uniform prior. So, I can check if MCMC can actually work.
The code below simulates n1, n2 and n3 with a particular λ value and does MCMC sampling to estimate λ’s posterior with simulated values, then outputs 5000 samples.
import sys
import numpy
import pymc
##simulated frequencies of triplets
l = 0.598 #true lambda = 0.598
#l = 0.162 #or lambda = 0.162
prob = [1-2*numpy.exp(-l)/3, numpy.exp(-l)/3, numpy.exp(-l)/3]
count_obs = numpy.random.multinomial(100, prob)
print(l, prob, count_obs)
##Bayesian model
lambd = pymc.Uniform("lambda", lower=0.0, upper=5.0) #Uniform prior for lambda
#A pymc function translating lambda to 3 probabilities of triplets
@pymc.deterministic
def triplet_prob(lambd=lambd):
p1 = 1-2*numpy.exp(-lambd)/3
p2 = p3 = numpy.exp(-lambd)/3
return [p1, p2, p3]
#observed values were associated with the multinomial model
obs = pymc.Multinomial("obs", n=sum(count_obs), p=triplet_prob, observed=True, value=count_obs)
#run MCMC
model = pymc.Model([obs, triplet_prob, lambd])
mcmc = pymc.MCMC(model)
mcmc.sample(100000, burn=50000)
with open("trace.lambda.%0.3f.txt"%l, "w") as f:
for i in mcmc.trace("lambda")[::10]:
f.write("%f\n"%i)
PyMC has a quite elegant and intuitive way to abstract a Bayesian modelling.
You can easily define prior distributions of parameters and develop models by combining them. The observed data are connected to the model with the “observed=True” option. Dependency of variables can be traced by “parent” and “children” attributes. Then, you can run MCMC just by calling mcmc.sample.
The distribution of posterior probability of λ when λ = 0.598 was like this. (In this case, simulated numbers are [n1,n2,n3]=[60,21,19])

The left plot is the trace of MCMC, and the right the histogram of MCMC samples. The curve superimposed on the histogram is an analytical solution. As you can see, the MCMC distribution fitted the analytical solution surprisingly well. This is great. The 95% credible interval (CI) is (0.30, 0.78). So, I am quite sure that λ is larger than zero and topology distribution is skewed.
When λ is smaller (λ = 0.162,[n1,n2,n3]=[44, 33, 23]), estimation became more uncertain. The 95%CI is (0.035, 0.38). A bit difficult to say n1 is more frequent.

I think this credible interval approach is OK for this simple case to just estimate λ. But, if you seriously implement species delimitation, a model comparison with reversible jump MCMC is required. It looks much more laborious to write codes for it since PyMC doesn’t have rjMCMC functions.
Regardless, I think PyMC is a good package which is easy to learn and have nice, readable documentations. It is probably a reasonable option if you want to integrate a Bayesian inference in your Python codes. Also, I think it is a handy tool to prototype a Bayesian model before writing it from scratch with other fast languages.
