As I test the tr2-delimitation with more data sets and read more papers applying it to difficult species, I find that it sometimes infers an unrealistically large number of species. These oversplits appear to happen more frequently when tr2 is used with very large data sets (like thousands of loci of RAD or RNA).
A reasonable explanation for this tendency of oversplit is tr2 delimits structured populations as species even if they are connected with gene flow. Whether molecular species delimitation methods actually delimits species or not is still under debate (like this paper). But, it is quite possible that tr2 falsely finds populations connected with weak gene flow as “species”.
The basic idea of tr2 delimitation is that sets of gene tree topology are more similar to each other when multiple species exist in your samples. This concordance of topology results in a skewed distribution of triplets, where one triplet topology is more frequently observed than other two.
Reduced gene flow between populations also creates topological concordance. The effect of reduced gene flow is usually much smaller than the effect of true speciation and often undetectable with a small number of loci. However, even a minimal skew of triplet distribution is detected as a signature of species when the number of loci is VERY large.
Because the tr2’s triplet distribution model only considers triplets’ topology and does not include the distribution of branch length, the gene flow likely has more significant effect on its performance than the full multi-species coalescent model (such as BPP).
I checked with simulations how reduced gene flow between populations affects the delimitation results of tr2.
Gene trees were simulated under a model where two populations split but retain genetic exchange with gene flow. The age of split, T, is nearly the same value of effective population size, Ne. For example, if Ne = 50,000 and generation time is 1 year, the time of split is 50,000 years ago. The amount of gene flow, Ne*m = 0.5, 1.0 and 5.0. In this simulation, gene tree’s topology is known without error. (I will consider situations with unresolved gene trees in future posts.)

Does tr2 split these two populations, pop.1 and pop.2, into separate species?
The plot below shows proportions of trials where samples from pop.1 and pop.2 are assigned to the same species and how the proportions change with the number of used loci. Different colors represent different degrees of gene flow.
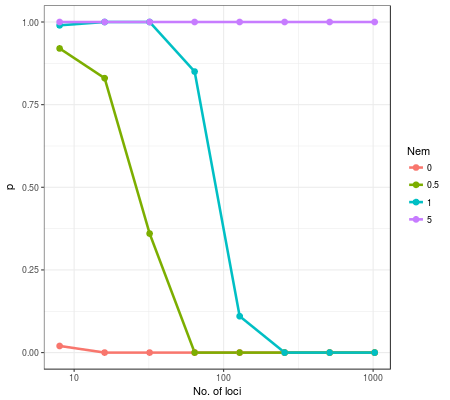
As you can see, even under conditions with moderate gene flow (Nem=0.5 or 1), samples from two populations were split into two spurious species. For example, populations connected with gene flow of Nem=1.0 were always split into two species when the number of loci exceeded ~500.
With Nem=5, which means gene flow is very large, the tr2 did not split populations even with 1000 loci (, but I guess it will probably oversplit if much larger number of inputs are used). When Nem=0, that is, the two populations are truely two young species, just about 10 loci was enough to detect them.
The reduced gene flow does have a strong effect on delimitation. So, you need to be careful when interpreting the results of multilocus delimitation with a very large data set. Splits detected only with hundreds or thousands of loci are probably not species, but population structure.
In the simulation above, only 10 or 20 loci with well-resolved tree topology have enough power to detect young species. Therefore, using different size of inputs and checking how patterns of split appear by increasing loci may help us interpret results.
Ultimately, it is quite hard to decide one threshold of gene flow and an appropriate sample size with which we can confidently say “there are species” since speciation process is continuous. Also, variation of informativeness of loci makes this decision even more difficult (You need more loci when they are less informative).
It seems that explicit modelling and quantification of gene flow is a better way to tackle this problem and should be a new direction of the multilocus delimitation program.

Thank you for sharing your findings. Are there plans to publish these results?