Installing Python and check versions
Python is required to run the tr2-delimitation on your computer. In many modern operating systems, Python is pre-installed and you do not need to install it by yourself.
However, the installed version of Python matters. The tr2 is written in Python 2, while, in some recent systems, Python 3 is the default version. This is simply because Python 2 was a standard when I started writing the codes, but Python 3 is becoming a new standard now. (I am translating the tr2 codes into Python3.) So, you first need to check the version of Python installed on your system.
Type on your console,
$python --version
If you see a message like below, you are running Python 2.
Python 2.7.6
In some decent OS’s, both Python2 and 3 are pre-installed, and you can call Python 2 by
$python2
even when your default Python is Python3.
If your environment does not have Python2 at all, visit the Python website and install it, or create a Python2 environment as explained in the next section.
Installing dependencies (scipy/numpy and Java)
Python packages called numpy/scipy is required to run tr2. These packages are for numerical calculations. Visit SciPy website and follow the instructions for your operating system to install Scipy libraries. Installing all Scipy related packages following the instruction is just fine though not all Scipy packages are required to run tr2.
An alternative, easy way to install dependencies is installing an all-in-one suite like Anaconda, which includes Python and related packages. As Anaconda allows you to run multiple versions of Python, you can install it with any version of Python.
If you choose to install Anaconda with Python3, you must run Python2 codes by creating Python2 environment.
$conda create --name python2 python=2 numpy scipy
This “conda” command creates an “environment” where the python version is 2 with numpy/scipy installed. You can switch to it by calling the “activate” command,
$activate python2
and switch back to the default environment by the “deactivate” command.
(python2) $deactivate
Checking packages
You can check if the packages are properly installed by loading them on the interactive shell.
$python
(Just by typing “python”, “interactive shell” starts. You can quit this mode by pressing Ctl+d.)
>>>import numpy, scipy
If it does not return errors, packages are ready to use.
Java is also required to run the consensus tree building program, triplec. Almost all modern operating systems have Java as pre-installed software.
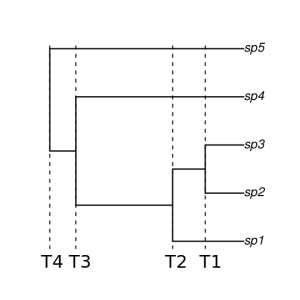

 One surprising thing is that ASTRAL was able to reconstruct a correct species tree even with apparently uninformative gene trees. It seems delimitation with tr2 can work correctly too once the species tree is correctly estimated.
One surprising thing is that ASTRAL was able to reconstruct a correct species tree even with apparently uninformative gene trees. It seems delimitation with tr2 can work correctly too once the species tree is correctly estimated.





{kind=link}
{kind=link}